Rare, complex, and potentially disfiguring, neurofibromatosis is not the kind of condition that we usually associate with plastic surgery. Neurofibromatosis is a genetic disorder characterized by the presence of neurofibromas — a series of tumors that grow on the brain, the spinal cord, or under the skin.
The type and severity of your neurofibromatosis will determine if you need more or less aggressive tactics to manage it. Managing neurofibromatosis may require an entire healthcare team, from a neurology expert to a reconstructive surgeon.
What are the causes of neurofibromatosis?
“Neurofibromatosis” really refers to three different but closely linked conditions that affect the nervous system. All three types are genetic, meaning they’re caused by the presence of specific genetic mutations.
You can inherit these gene mutations from your parents or grandparents, or they can appear randomly not long after conception.
These mutations affect the gene that produces neurofibromin, a protein that controls the growth of new cells in your nervous tissue. Without enough neurofibromin, your nerves and spinal cord may grow too quickly and form tumors around the body.
In some cases, neurofibromatosis can be relatively mild, even if it requires constant monitoring. Other times, it can cause hearing loss, heart problems, learning disabilities, blindness, or severe pain.
What determines how severe your condition is? Mainly luck. Neurofibromatomas are typically benign, but depending on where they develop, they can damage many surrounding nerves.
What are the risk factors for neurofibromatosis?
As with any genetic condition, the most important risk factor lies in your family history. If any of your parents have the disease, you’ll have a 50% chance of developing it too. It is regarded as an autosomal dominant disease, so only one of your parents needs to have it for you to be at risk.
In addition, if anyone in your extended family (aunts, uncles, grandparents, etc.) has the condition, you may also be more likely of developing a “spontaneous” gene defect.
But other than that? We don’t know. All neurofibromatosis types are remarkably rare, affecting between 1 in 3,000 to 25,000 babies born. Experts haven’t identified any particular ethnicity, group, exposure to a chemical, or anything else that increases the risk of the disease.
What are the symptoms of neurofibromatosis?
Currently, we use the term “neurofibromatosis” for three separate diseases, each one affecting a different gene. The first two, so far, seem to be deeply intertwined and often difficult to tell apart. The third one still leaves some room for speculation.
Neurofibromatosis Type 1 (NF 1)
Also known as von Recklinghausen disease, NF1 happens due to a mutation in the NF1 gene, which is located on chromosome 17. According to the National Institute of Neurological Disorders and Stroke, NF1 usually appears during early childhood, and it’s almost always evident by the age of 10.
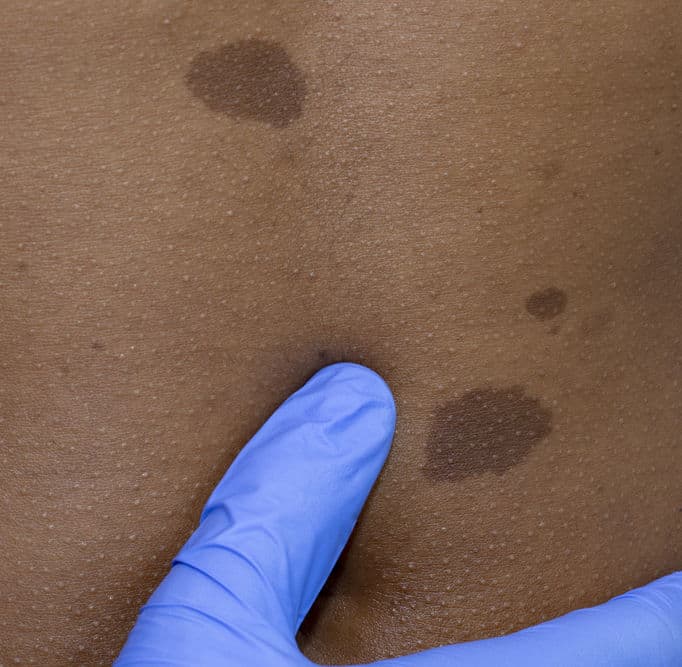
Usually, the first symptom to appear is cafe au lait spots. These are light brown spots (the color of a dark latte), that appear around the body during the first two years of life. They’re totally harmless by themselves, and they may seem like a completely normal birthmark. However, if you (or your child) have more than six, they’re over 15 millimeters long each, and have irregular edges, then they may indicate NF1. At the very least, a pediatrician will suggest keeping a close eye on a baby with these spots.
Other neurofibromatosis type 1 symptoms include:
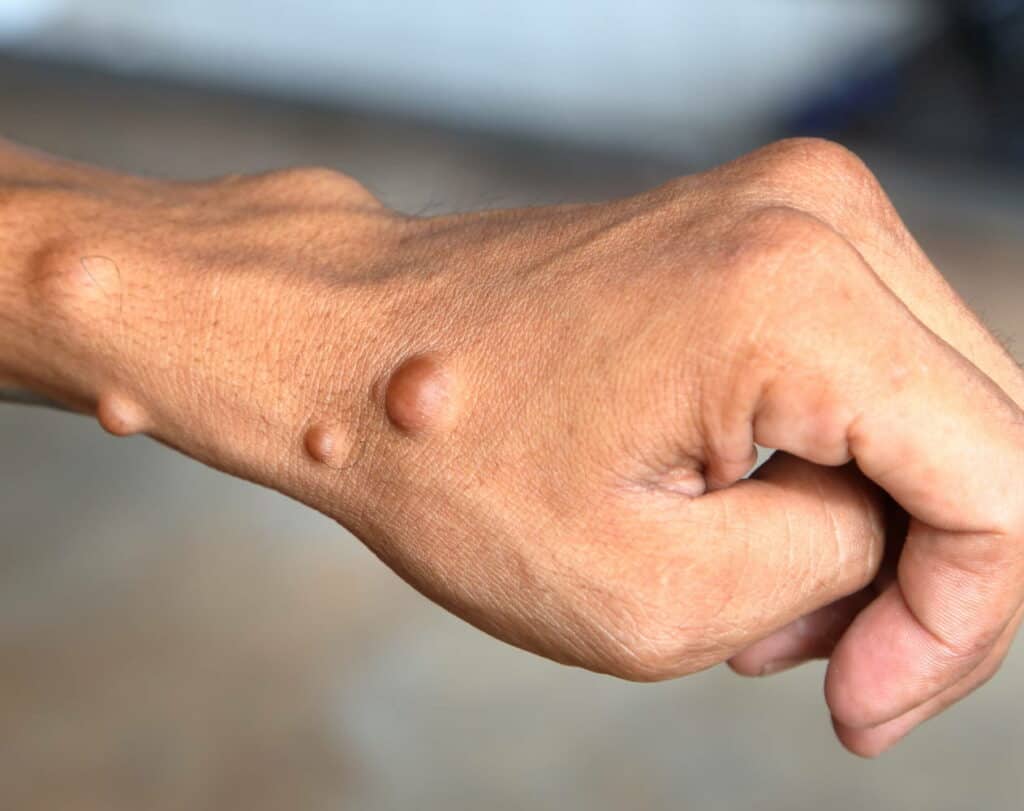
- Many cutaneous neurofibromas, or small pea-sized bumps in the skin.
- Plexiform neurofibromas, or one large tumor impacting several nerves, usually during adolescence.
- Freckles in the armpit or groin, usually before the age of five.
- Two or more Lisch nodules, or tiny growths on the iris of the eye.
- Benign tumors or gliomas on the auditory or optic nerves (optic gliomas).
- Bone problems such as scoliosis, deformities in the eye socket bone, or tibia (the long bone in the shin) abnormalities.
Children with NF1 are often at an increased risk of many developmental issues, from ADHD (attention deficit hyperactivity disorder) to learning disabilities. Depending on where the tumors develop, they can also develop:
- Blindness
- Frequent fractures
- Cardiovascular problems, especially high blood pressure
- Short stature
- Brain tumors
Neurofibromatosis 1 is the most common type of neurofibromatosis and affects 1 in every 3,000 children. People with NF1 have a shortened life expectancy of about 54 years.
Neurofibromatosis Type 2 (NF2)
Much rarer than NF1, type 2 neurofibromatosis affects the NF2 gene on chromosome 22. This gene affects both neurofibromin production and merlin production — a second protein that also suppresses tumors. Many kids with NF2 are not diagnosed until they’re in their early- to mid-teens.
Typical symptoms include schwannomas, or tumors made up of Schwann cells. These cells produce myelin, which in turn protects several important nerve endings. Schwannomas usually appear on the cranial nerves, especially on the vestibular nerve (vestibular schwannomas) or the acoustic nerve (acoustic neuromas). The vestibular nerve connects to the inner ear and plays an important role in balance and hearing. Usually, these balance problems are what “uncovers” the disorder.
Other possible NF2 symptoms include:
- Skin schwannomas, which may look like bumps under the skin.
- Meningiomas, or benign tumors in the meninges that surround the brain and spinal cord.
- Ependymomas, or asymptomatic tumors on the spinal cord.
- Depending on the location of the tumors, people with NF2 have a higher chance of developing hearing and vision problems, cataracts, muscle weakness, or seizures. All the typical tumors we mentioned are benign, but occasionally, they can turn cancerous.
Schwannomatosis
This is the rarest type of neurofibromatosis, and some neurologists believe it to be completely separate. We are not sure exactly what causes it, but it seems to involve different genes than NF1 or NF2.
Schwannomatosis often appears very similar to NF2, with the same combination of slow-growing schwannomas and meningiomas. However, it tends to appear later in life, sometimes even after the age of 30. People with schwannomatosis are also more likely to suffer from chronic pain, but less likely for their tumors to turn cancerous.
Still, telling schwannomatosis and NF2 apart is remarkably difficult — unless you already have an established family history, genetic testing may be the only way to tell them apart for sure.
How do we treat neurofibromatosis?
For people with neurofibromatosis, their quality of life will depend largely on which nerve cells are affected by the tumors, and on whether they turn cancerous. This means treatment can vary wildly.
Can neurofibromatosis be cured?
The short answer is no. However, it can be managed, depending on when you’re first diagnosed and how it evolves. Some options include:
- For kids with NF1, a drug known as selumetinib can help prevent new tumors from growing.
- Surgery to correct the curvature of the spine.
- Surgical extraction or chemotherapy for optic gliomas.
- An auditory brain stem implant to restore hearing.
- Pain management, if any of the tumors start pressing on peripheral nerve tissue.
Most people will also need frequent check-ups, and even occasional MRI scans to monitor the growth of their tumors or to spot new ones.
What can a plastic surgeon do for me?
Even in the best-case scenario, neurofibromatosis can have a deep impact on your self-image and self-esteem.
Depending on the type of tumor and the parts of the body affected, we will use one of two options:
- A regular surgical excision for anything subcutaneous (under the skin). In these cases, we will have to be extra careful, as we can’t see which nerves are involved from the outside. We’ll need an MRI or ultrasound scan before we can plan the surgery properly.
- For superficial skin schwannomas, we can remove a large number of lesions using electrodesiccation — drying the skin tissue without burning.
If the tumors have been growing rapidly, we’ll also need to send them to a pathology lab to rule out a malignancy.
Looking for a neurofibromatosis surgeon near LA? Call Dr. Saber
Neuromatosis is the kind of complex condition that can humble any single specialist. For patients who are looking to deal with its more visible signs, it is extremely important to work with an experienced clinician who understands its nuances and isn’t afraid to ask for help.
Dr. Sepideh Saber is a board-certified plastic and reconstructive surgeon. Her combined training, which spans from Stanford University to New York University, totals more than 10 years in reconstructive procedures and microsurgery. In addition, her philosophy of “health partnerships” and open communication offer a more empathetic approach for people of all ages dealing with major life changes or complex conditions.
To get in touch, call (877) 205-4100 or schedule a consultation online.
The practice of Dr. Saber is located in Encino, CA for patients throughout the Los Angeles area. We are also convenient to Encino, Woodland Hills, Sherman Oaks, Calabasas, Burbank, Glendale, Hidden Hills, Agoura Hills, Northridge, North Hollywood, Malibu, Topanga, Canoga Park, Reseda, Valley Glen, Chatsworth, West Hills, Winnetka, Universal City, Bel Air, Beverly Hills, Downtown Los Angeles, Silverlake, and Echo Park.
Frequently asked questions about neurofibromatosis
How is neurofibromatosis diagnosed?
Often, the most important “red flag” for Neurofibromatosis is having a parent with it.
For NF1, cafe au lait spots may serve as a clue. However, the National Institutes of Health (NIH) suggests either genetic testing, as well as detailed imaging of the brain before a definitive diagnosis.
Does neurofibromatosis hurt?
This depends largely on where tumors develop. If a tumor starts pressing on the brain stem or on another organ, then yes, it can be very painful. In addition, people with schwannomatosis often experience chronic pain, even if none of their nerves are visibly involved.
Is neurofibromatosis deadly?
It usually isn’t — but if any of the tumors turn malignant, it can be. This is why specialists recommend watching out for any tumor that grows too rapidly — and why I insist on sending anything to the pathologist.
What can I do to help people with neurofibromatosis?
What we now need most of all is more research. If you’re interested in donating or organizing awareness events, contact the Children’s Tumor Foundation.
See MORE of Dr. Saber’s transformations!
Explore other Before & After galleries...
What patients say about Dr. Saber …
Stories from Our Blog We Think You’ll Like
see more articles from our blog...